жө…и°Ҳпјҡз«ӢејӮиҚҜзү©INDз”іиҜ·д№ӢиҚҜеӯҰз ”з©¶
з«ӢејӮиҚҜзү©зҡ„ејҖеҸ‘жҳҜдёҖдёӘжһҒе…·жҺўзҙўжҖ§зҡ„з ”з©¶еҺҶзЁӢ�пјҢпјҢ�пјҢпјҢ�пјҢе…¶йҖҡеёёз”ұжңӘзҹҘжңҖе…Ҳ�пјҢпјҢ�пјҢпјҢ�пјҢеҹәдәҺжңӘиў«зҹҘи¶ізҡ„дёҙеәҠйңҖжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢеҺ»ејҖеұ•иҚҜзү©зӯӣйҖүдёҺеҸ‘жҳҺзҡ„дәӢжғ…�гҖӮ�гҖӮ�гҖӮгҖӮе·®еҲ«дәҺд»ҝеҲ¶иҚҜ�пјҢпјҢ�пјҢпјҢ�пјҢз«ӢејӮиҚҜзү©зҡ„з ”з©¶�пјҢпјҢ�пјҢпјҢ�пјҢжҳҜйҡҸзқҖе·®еҲ«йҳ¶ж®өиҖҢйҖҗжӯҘж·ұе…ҘзқҒејҖзҡ„�пјҢпјҢ�пјҢпјҢ�пјҢжҜҸйҳ¶ж®өз ”з©¶зҡ„ж·ұеәҰйҖҡ�пјӣ�пјӣ�пјӣ�пјӣеІ«жқӮПҲз•”з…ҠОҰзәіз¬ғе§ҘеіЎзЎӯ�пјҢпјҢ�пјҢпјҢ�пјҢд»ҺиҖҢйҳ»жӯўдёҚйЎ»иҰҒзҡ„еӨӘиҝҮејҖеҸ‘�пјҢпјҢ�пјҢпјҢ�пјҢд»ҘиҠӮзәҰеҗ„ж–№йқўзҡ„иө„жәҗ�гҖӮ�гҖӮ�гҖӮгҖӮеңЁжҲ‘еӣҪ�пјҢпјҢ�пјҢпјҢ�пјҢз«ӢејӮиҚҜзү©зҡ„ејҖеҸ‘�пјҢпјҢ�пјҢпјҢ�пјҢиҷҪиө·жӯҘиҫғжҷҡдё”е°ҡдёҚеҸҜзҶҹ�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶж•ҙдҪ“и¶ӢеҠҝжӯЈеңЁиҝҪиө¶иҘҝ欧ж—Ҙ�пјҢпјҢ�пјҢпјҢ�пјҢжҜҸдёӘзҺҜиҠӮз»ҶиҠӮд№ҹйғҪеңЁе®Ңе–„еҪ“дёӯ�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮдёӢйқўиҰҒиҒҠзҡ„INDз”іиҜ·дёӯзҡ„вҖңиҚҜеӯҰз ”з©¶вҖқ�пјҢпјҢ�пјҢпјҢ�пјҢе°ұжҳҜе…¶дёӯд№ӢдёҖ�гҖӮ�гҖӮ�гҖӮгҖӮ
зҫҺеӣҪFDA&з«ӢејӮиҚҜINDйҳ¶ж®ө-иҚҜеӯҰз ”з©¶
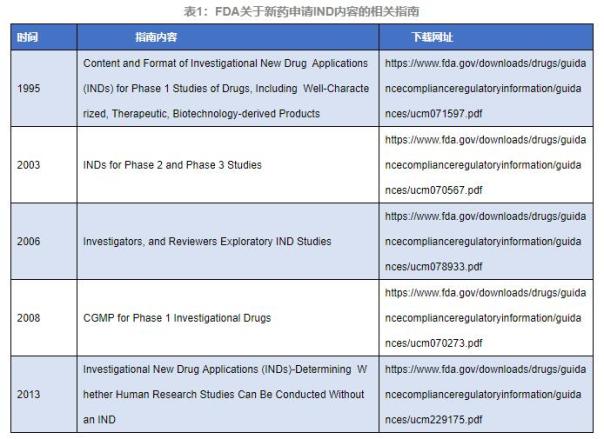
1. FDAе…ідәҺINDйҳ¶ж®өиҚҜзү©зҡ„е®ЎиҜ„жҢҮеҚ—
ж—©еңЁ1995е№ҙ�пјҢпјҢ�пјҢпјҢ�пјҢзҫҺеӣҪйЈҹзү©иҚҜе“Ғзӣ‘и§ҶжІ»зҗҶеұҖзҡ„CDERе’ҢCBERдҫҝе®ЈеёғдәҶе…ідәҺINDйҳ¶ж®өзҡ„иҚҜзү©жҢҮеҚ—вҖңContent and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived ProductвҖқ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҘжҢҮеҚ—еү–жһҗдәҶ21 CFRдёӯ312.22е’Ң312.23е…ідәҺжңҖеҲқиҝӣе…ҘзҫҺеӣҪдёҙеәҠз ”з©¶иҜ•йӘҢиҚҜзү©зҡ„ж•°жҚ®иҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢе…Ғи®ёINDйҳ¶ж®өжҸҗдәӨзҡ„з§Қз§Қз ”з©¶зҡ„ж•°жҚ®еҸҠж·ұеәҰ�пјҢпјҢ�пјҢпјҢ�пјҢе…·жңүжһҒеӨ§зҡ„ж— йӮӘжҖ§�гҖӮ�гҖӮ�гҖӮгҖӮ
2003е№ҙ�пјҢпјҢ�пјҢпјҢ�пјҢFDAеҸҲе®ЈеёғдәҶжҢҮеҚ—вҖңINDs for Phase 2 and Phase 3 StudiesвҖқ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҘжҢҮеҚ—иҝӣдёҖжӯҘжҸҗдҫӣз”іиҜ·дәә(INDйҳ¶ж®ө)е…ідәҺIIжңҹ/IIIжңҹдёҙеәҠеҢ–еӯҰгҖҒз”ҹдә§гҖҒCMCдҝЎжҒҜзӯүеҶ…е®№зҡ„зӣёе…іе»әеӘҫе’ҢиҰҒжұӮ�гҖӮ�гҖӮ�гҖӮгҖӮ
2008е№ҙ�пјҢпјҢ�пјҢпјҢ�пјҢFDAе®ЈеёғжҢҮеҚ—вҖңCGMP for Phase 1 Investigational DrugsвҖқ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҘжҢҮеҚ—иҜҰз»ҶеҸҷиҝ°дәҶв… жңҹдёҙеәҠиҜ•йӘҢж ·е“Ғз”ҹдә§иҗҪе®һcGMPзҡ„й—®йўҳ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®жҺҘзәіиҚҜзү©иҙЁйҮҸжҺ§еҲ¶(QC)еҺҹеҲҷдҪңдёәcGMPзҡ„дёҖйғЁеҲҶ�пјҢпјҢ�пјҢпјҢ�пјҢд»ҺиҖҢеҢ…з®Ўв… жңҹдёҙеәҠиҜ•йӘҢж ·е“Ғзҡ„иҙЁйҮҸе’Ңжё…йқҷжҖ§�гҖӮ�гҖӮ�гҖӮгҖӮ
2013е№ҙ�пјҢпјҢ�пјҢпјҢ�пјҢFDAеҶҚж¬Ўе®ЈеёғжҢҮеҚ—вҖңInvestigational New Drug Applications (INDs) - Determining Whether Human Research Studies Can Be Conducted Without an INDвҖқ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҘжҢҮеҚ—ж—ЁеңЁиө„еҠ©дёҙеәҠз”іиҜ·дәәзЎ®е®ҡж–°иҚҜINDз”іиҜ·дёӢ�пјҢпјҢ�пјҢпјҢ�пјҢж¶үеҸҠзӣёе…ізҡ„з ”з©¶жҳҜеҗҰеҝ…йңҖиў«з ”з©¶�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮ21 CFR 312йғЁеҲҶ�пјӣ�пјӣ�пјӣ�пјӣ并иҜҰз»ҶиҜҙжҳҺжҷ°дҪ•ж—¶йңҖиҰҒINDз”іиҜ·�пјҢпјҢ�пјҢпјҢ�пјҢдҪ•з§Қжғ…еҪўдёҚйңҖиҰҒINDз”іиҜ·�пјҢпјҢ�пјҢпјҢ�пјҢзЎ®е®ҡдәҶдёҖе®ҡзҡ„йҖӮ用规模�гҖӮ�гҖӮ�гҖӮгҖӮ
2. INDиҚҜеӯҰйғЁеҲҶ-еҲҶеқ—з»Ҷиҝ°
еҮӯиҜҒзҫҺеӣҪFDAе®Јеёғзҡ„зӣёе…іжҢҮеҚ—зҡ„иҜҰз»ҶеҶ…е®№�пјҢпјҢ�пјҢпјҢ�пјҢеңЁе·®еҲ«дёҙеәҠз”іжҠҘйҳ¶ж®ө�пјҢпјҢ�пјҢпјҢ�пјҢе…¶еҜ№з«ӢејӮиҚҜзү©-иҚҜеӯҰйғЁеҲҶзҡ„иҙЁж–ҷиҚҜгҖҒзЁіеӣәжҖ§гҖҒиҙЁйҮҸгҖҒеҲ¶еүӮзӯүеҶ…е®№�пјҢпјҢ�пјҢпјҢ�пјҢе…·жңүиҜҰз»Ҷзҡ„еҲҶж®өз ”з©¶иҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҰжғ…еҰӮдёӢпјҡ
вҳҶиҙЁж–ҷиҚҜйғЁеҲҶ
вҳҶвҳҶIжңҹдёҙеәҠ
еҲ¶еӨҮе·Ҙиүә~жҸҗдҫӣеҗҲжҲҗе·Ҙиүәз ”з©¶зҡ„з®ҖиҰҒжҖ»з»“�пјҢпјҢ�пјҢпјҢ�пјҢиҜҙжҳҺзҺ°жңүиҜ•еҲ¶и§„жЁЎ�пјҢпјҢ�пјҢпјҢ�пјҢеҗҲжҲҗи№Ҡеҫ„еӣҫдёӯе»әи®®жҳҺзЎ®еҗ„еҠһжі•зҡ„еҸҚеә”жқЎд»¶гҖҒжүҖз”ЁиҜ•еүӮгҖҒжә¶еүӮгҖҒеӮ¬еҢ–еүӮзӯү�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®жңҖе…Ҳе…іжіЁеҜ№иҰҒе®іиө·е§ӢиҙЁж–ҷзҡ„иҙЁйҮҸж•°жҚ®з§ҜзҙҜ�пјӣ�пјӣ�пјӣ�пјӣеӣ зІҫеҲ¶е·Ҙиүәзҡ„е·®еҲ«еҸҜиғҪеҪұе“Қдә§е“Ғзҡ„жқӮиҙЁи°ұгҖҒжҷ¶еһӢгҖҒзІ’еәҰзӯү�пјҢпјҢ�пјҢпјҢ�пјҢйңҖжіЁйҮҚиҜҙжҳҺзІ—е“Ғзҡ„зәҜеҢ–/зІҫеҲ¶иҰҒйўҶ�гҖӮ�гҖӮ�гҖӮгҖӮ
зү№еҫҒеҲӨж–ӯ~жӯӨйҳ¶ж®өжҸҗдҫӣж”ҜжҢҒеҢ–еӯҰз»“жһ„зҡ„иө·жәҗз ”з©¶ж•°жҚ®еҚіеҸҜ�пјӣ�пјӣ�пјӣ�пјӣиҜҙжҳҺеҸҜиғҪеҪұе“Қжё…йқҷжҖ§зҡ„зҗҶеҢ–жҖ§еӯҗ�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮж¶ҲиһҚжҖ§(е·®еҲ«pHжә¶ж¶Ідёӯ)гҖҒзІ’еәҰгҖҒжҷ¶еһӢзӯү�гҖӮ�гҖӮ�гҖӮгҖӮе»әи®®еңЁж—©жңҹдёҙеәҠйҳ¶ж®өеҚізЎ®е®ҡиҚҜз”Ёжҷ¶еһӢ�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶзІ’еәҰиҝҳйңҖиҰҒиҝһзі»дёҙеәҠз ”з©¶зҡ„жҺЁиҝӣдёҖзӣҙз§ҜзҙҜж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶII/IIIжңҹдёҙеәҠ
еҲ¶еӨҮе·Ҙиүә~жҸҗдәӨеҲ¶еӨҮе·Ҙиүәзҡ„иҪ¬еҸҳеҸҠзӣёе…із ”究иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢиҜ„дј°еҸҳжҚўеҜ№дә§е“Ғзҡ„иҙЁйҮҸе’Ңжё…йқҷжҖ§зҡ„еҪұе“Қ�пјӣ�пјӣ�пјӣ�пјӣе…ідәҺеҢ…з®Ўдә§е“Ғжё…йқҷжҖ§зҡ„з”ҹдә§еҠһжі•(еҰӮеҸ‘й…өдә§е“Ғзҡ„зәҜеҢ–еҠһжі•)зҡ„еҺҶзЁӢжҺ§еҲ¶еә”жңүжё…жҷ°еҪўиІҢ�пјӣ�пјӣ�пјӣ�пјӣжҸҗдҫӣиө·е§ӢиҙЁж–ҷзҡ„иҙЁйҮҸжҺ§еҲ¶дҝЎжҒҜ(жіүжәҗгҖҒеү–жһҗиҰҒйўҶгҖҒжЈҖжөӢж•Ҳжһң)�пјҢпјҢ�пјҢпјҢ�пјҢе…ідәҺз»“жһ„йҮҚеӨ§зҡ„иҰҒе®іиө·е§ӢиҙЁж–ҷеә”жҸҗдҫӣиҜҰз»Ҷз”ҹдә§е·ҘиүәдҝЎжҒҜ�пјӣ�пјӣ�пјӣ�пјӣжҸҗдҫӣиҰҒе®іеҠһжі•гҖҒдёӯеҝғдҪ“зҡ„жҺ§еҲ¶дҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮ
зү№еҫҒеҲӨж–ӯ~жҸҗдҫӣеҗҲзҗҶж”ҜжҢҒиҚҜзү©еҢ–еӯҰз»“жһ„зҡ„иҜҒе®һ�пјҢпјҢ�пјҢпјҢ�пјҢеҚ•жҷ¶XиЎҚе°„гҖҒжһ„иұЎеү–жһҗзӯүеҸҜеңЁIIIжңҹжҸҗдҫӣ�пјӣ�пјӣ�пјӣ�пјӣиҝһзі»дёҙеәҠиҜ•йӘҢеҲ¶еүӮзҡ„еүӮеһӢзү№зӮ№е’ҢиҚҜзү©зү№еҫҒ�пјҢпјҢ�пјҢпјҢ�пјҢжҸҗдҫӣиҝӣдёҖжӯҘе®Ңе–„зҡ„иҙЁж–ҷиҚҜзҗҶеҢ–жҖ§еӯҗдҝЎжҒҜ�пјҢпјҢ�пјҢпјҢ�пјҢеҢ…жӢ¬ж¶ҲиһҚжҖ§гҖҒжҷ¶еһӢгҖҒзІ’еәҰгҖҒжё—йҖҸжҖ§гҖҒж—Ӣе…үжҖ§гҖҒеј•ж№ҝжҖ§гҖҒеҲҶжҙҫзі»ж•°гҖҒз”өзҰ»еёёж•°зӯү�пјҢпјҢ�пјҢпјҢ�пјҢе…ідәҺеҸЈжңҚеӣәдҪ“еҲ¶еүӮ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®е°Ҫж—©з ”з©¶е…¶иҙЁж–ҷиҚҜзҡ„жё—йҖҸжҖ§�пјҢпјҢ�пјҢпјҢ�пјҢзӣёиҜҶе…¶BCSеҲҶзұ»�пјҢпјҢ�пјҢпјҢ�пјҢеҜ№еҲ¶еүӮеӨ„ж–№е·ҘиүәејҖеҸ‘д»ҘеҸҠдҪ“еӨ–йҮҠж”ҫиҰҒйўҶзҡ„е»әи®ҫеҫҲжңүиө„еҠ©�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶзЁіеӣәжҖ§з ”究
вҳҶвҳҶIжңҹдёҙеәҠ
жҸҗдҫӣе·Іжңүзҡ„зЁіеӣәжҖ§иҜ•йӘҢж•ҲжһңгҖҒеҗҺз»ӯзҡ„зЁіеӣәжҖ§з ”究еҰ„жғі�пјӣ�пјӣ�пјӣ�пјӣе…ідәҺеӨҚжә¶гҖҒзЁҖйҮҠжҲ–ж··ж·ҶеҗҺеӨҡж¬Ўеә”з”Ёзҡ„еҲ¶еүӮ�пјҢпјҢ�пјҢпјҢ�пјҢеә”ејҖеұ•дҪҝз”Ёдёӯзҡ„зЁіеӣәжҖ§з ”究�пјӣ�пјӣ�пјӣ�пјӣе»әи®®ејҖеұ•еҪұе“Қеӣ зҙ зӯүиҜ•йӘҢ�пјҢпјҢ�пјҢпјҢ�пјҢд»ҘзӣёиҜҶиҚҜзү©зҡ„еҶ…еңЁзЁіеӣәжҖ§жғ…еҪўгҖҒжҪңеңЁзҡ„йҷҚи§ЈйҖ”еҫ„�пјҢпјҢ�пјҢпјҢ�пјҢиө„еҠ©зЁіеӣәжҖ§иҜ•йӘҢжқЎд»¶зҡ„йҖүжӢ©гҖҒеү–жһҗиҰҒйўҶзҡ„иҖғеҜҹ�гҖӮ�гҖӮ�гҖӮгҖӮе·Іжңүзҡ„зЁіеӣәжҖ§з ”究ж•Ҳжһңеә”ж”ҜжҢҒжӢҹдёҫиЎҢзҡ„дёҙеәҠз ”з©¶�пјҢпјҢ�пјҢпјҢ�пјҢеҢ…з®ЎеҲ¶и®ўдёҙеәҠиҜ•йӘҢж—¶д»ЈиҚҜе“ҒиҙЁйҮҸзҡ„зЁіеӣә�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶII/IIIжңҹдёҙеәҠ
жҖ»з»“е·ІиҺ·еҫ—д»ЈиЎЁжҖ§жү№ж¬Ўзҡ„зЁіеӣәжҖ§иҜ•йӘҢж•°жҚ®�пјӣ�пјӣ�пјӣ�пјӣеҪўиІҢиҙЁж–ҷиҚҜеҢ–еӯҰе’Ңзү©зҗҶж•Ҹж„ҹжҖ§�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮе…үж•Ҹж„ҹжҖ§гҖҒеҗёж№ҝжҖ§зӯү�пјҢпјҢ�пјҢпјҢ�пјҢжҪңеңЁзҡ„йҷҚи§ЈйҖ”еҫ„�гҖӮ�гҖӮ�гҖӮгҖӮIгҖҒIIжңҹдёҙеәҠиҜ•йӘҢйҖҡеёёе‘Ёжңҹиҫғй•ҝ�пјҢпјҢ�пјҢпјҢ�пјҢиҖҢжӢҹз”ЁдәҺдёҙеәҠиҜ•йӘҢж ·е“Ғзҡ„зЁіеӣәжҖ§иҖғеҜҹж—¶й—ҙеҫҲжңүйҷҗ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®еҸҜжҸҗдәӨзӣёе…ізҡ„ж”ҜжҢҒжҖ§з ”究数жҚ®�пјҢпјҢ�пјҢпјҢ�пјҢдҫӢеҰӮдёҙеәҠеүҚжҲ–ж—©жңҹдёҙеәҠиҜ•йӘҢзҡ„еӨ„ж–№гҖҒе·Ҙиүәзӣёдјјжү№ж¬Ўд»ҘеҸҠжү№йҮҸиҫғе°Ҹжү№ж¬Ўзӯүзҡ„зЁіеӣәжҖ§з ”究ж•Ҳжһң�гҖӮ�гҖӮ�гҖӮгҖӮиҝӣе…ҘIIIжңҹдёҙеәҠеҗҺйҖҡеёёеә”еҮӯиҜҒжҢҮеҜјеҺҹеҲҷиҰҒжұӮејҖеұ•зЁіеӣәжҖ§иҜ•йӘҢ�пјҢпјҢ�пјҢпјҢ�пјҢд»ҘеҲ©дҫҝNDAзҡ„з”іжҠҘ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶиҙЁйҮҸйғЁеҲҶ
вҳҶвҳҶIжңҹдёҙеәҠ
еҲ—еҮәиҙЁйҮҸж ҮеҮҶзҡ„йЎ№зӣ®гҖҒиҰҒйўҶе’ҢеҸҜжҺҘеҸ—йҷҗеәҰ�гҖӮ�гҖӮ�гҖӮгҖӮе»әи®®еҜ№ж¶үеҸҠжё…йқҷжҖ§зҡ„жңүе…ізү©иҙЁгҖҒйҒ—дј жҜ’жҖ§жқӮиҙЁзӯүжЈҖжөӢиҰҒйўҶзҡ„йҖӮз”ЁжҖ§дёҫиЎҢиө·жәҗйӘҢиҜҒ�пјҢпјҢ�пјҢпјҢ�пјҢиө·жәҗз•Ңе®ҡжқӮиҙЁи°ұ�пјӣ�пјӣ�пјӣ�пјӣеҲ¶и®ўйҷҗеәҰеә”еҹәдәҺе·Іжңүжү№еү–жһҗж•°жҚ®зҡ„з§ҜзҙҜ�пјҢпјҢ�пјҢпјҢ�пјҢдёҙеәҠж ·е“Ғзҡ„жқӮиҙЁж°ҙе№ідёҚеҫ—еҮҢй©ҫеҠЁзү©жё…йқҷжҖ§иҜ•йӘҢж•°жҚ®жүҖж”ҜжҢҒзҡ„е“Қеә”жқӮиҙЁзҡ„ж°ҙе№і�пјӣ�пјӣ�пјӣ�пјӣжҸҗдҫӣе·Іжңүжү№ж¬Ў(еҰӮжё…йқҷжҖ§иҜ„д»·гҖҒзЁіеӣәжҖ§иҜ•йӘҢзӯү)е’ҢжӢҹдёҫиЎҢдёҙеәҠиҜ•йӘҢжү№ж¬Ў(иӢҘжңү)зҡ„жү№еү–жһҗж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶII/IIIжңҹдёҙеәҠ
жҸҗдҫӣеү–жһҗиҰҒйўҶйғЁеҲҶйӘҢиҜҒж•Ҳжһңж‘ҳиҰҒ(еҸҜеҲ—иЎЁ�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮдё“еұһжҖ§гҖҒз»ҶеҜҶеәҰгҖҒеҮҶзЎ®еәҰгҖҒзәҝжҖ§гҖҒе®ҡйҮҸйҷҗ/жЈҖжөӢйҷҗзӯү)�пјӣ�пјӣ�пјӣ�пјӣ继з»ӯдёҫиЎҢжқӮиҙЁи°ұзҡ„еҲӨж–ӯ�пјӣ�пјӣ�пјӣ�пјӣеҜ№иҜҒж–ҷиҚҜеҗҲжҲҗе·ҘиүәеҸҳжҚўзҲҶеҸ‘зҡ„ж–°жқӮиҙЁе’ҢеҲ¶еүӮдёӯж–°еҸ‘жҳҺзҡ„йҷҚи§Јдә§е“ҒдёҫиЎҢе®ҡжҖ§е’Ңе®ҡйҮҸз ”з©¶�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®з”іжҠҘIгҖҒIIжңҹдёҙеәҠж—¶зЎ®е®ҡиҙЁж–ҷиҚҜдё»иҰҒжқӮиҙЁд»ҘеҸҠеҲ¶еүӮзҡ„дё»иҰҒйҷҚи§Јдә§е“Ғ�пјӣ�пјӣ�пјӣ�пјӣйҮҚж–°иҜ„дј°е…ҲеүҚIжңҹжҲ–IIжңҹзҡ„иҙЁйҮҸж ҮеҮҶе’ҢеҸҜжҺҘеҸ—йҷҗеәҰ�пјҢпјҢ�пјҢпјҢ�пјҢеҮӯиҜҒзӣ®д»Ҡзҡ„з ”з©¶йҳ¶ж®өиҝӣдёҖжӯҘиҜ„дј°е’Ңи°ғи§Ј�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺйҡҫжә¶жҖ§еҸЈжңҚеӣәдҪ“еҲ¶еүӮ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®з§ҜзҙҜеҲ¶еүӮжүҖз”ЁиҙЁж–ҷиҚҜзҡ„зІ’еәҰжј«иЎҚж•°жҚ®�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®ҫиҚҜзү©ејҖеҸ‘ж—©жңҹгҖҒеҗҺжңҹиҺ·еҫ—ж•°жҚ®дёҺдҪ“еҶ…з–—ж•Ҳзҡ„зӣёе…іжҖ§�пјӣ�пјӣ�пјӣ�пјӣе»әи®ҫжә¶еҮәеәҰ/йҮҠж”ҫеәҰиҰҒйўҶ�пјҢпјҢ�пјҢпјҢ�пјҢиҝһзі»иҚҜзү©зү№еҫҒйҖүжӢ©д»ӢиҙЁе’ҢиҜ•йӘҢиҰҒйўҶ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®еҜ№дёҙеәҠеүҚиҜ•йӘҢж ·е“ҒгҖҒеҗ„жңҹдёҙеәҠиҜ•йӘҢж ·е“ҒгҖҒзЁіеӣәжҖ§иҜ•йӘҢж ·е“Ғзҡ„жә¶еҮә/йҮҠж”ҫиЎҢдёәдёҫиЎҢиҖғеҜҹ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®ҫиҚҜзү©ејҖеҸ‘ж—©жңҹгҖҒеҗҺжңҹиҺ·еҫ—ж•°жҚ®дёҺдҪ“еҶ…з–—ж•Ҳзҡ„зӣёе…іжҖ§�гҖӮ�гҖӮ�гҖӮгҖӮжҸҗдәӨеҗ„йЎ№дёҙеәҠиҜ•йӘҢж ·е“Ғзҡ„жү№еү–жһҗж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶеҲ¶еүӮйғЁеҲҶ
вҳҶвҳҶIжңҹдёҙеәҠ
йҖҡеёёжҺҘзәізҡ„еүӮеһӢиҫғйҮҸз®Җжңҙ�пјҢпјҢ�пјҢпјҢ�пјҢдҫӢеҰӮеҸЈжңҚеҲ¶еүӮжҺҘзәізІүжң«иЈ…иғ¶еӣҠ�пјҢпјҢ�пјҢпјҢ�пјҢжҲ–иҖ…еҲ¶еӨҮжҲҗж··жӮ¬ж¶ІгҖҒжә¶ж¶Ізӯү�пјҢпјҢ�пјҢпјҢ�пјҢд»ҘеҲ©дҫҝеүӮйҮҸжҺўзҙў�пјҢпјҢ�пјҢпјҢ�пјҢжӯӨйҳ¶ж®өзҡ„еүӮеһӢе’ҢеӨ„ж–№е·ҘиүәиҝҳдҝқеӯҳеҫҲеӨ§зҡ„дёҚзЎ®е®ҡжҖ§�пјҢпјҢ�пјҢпјҢ�пјҢдёҚжҳҜиҚҜеӯҰиҜ„д»·зҡ„йҮҚзӮ№�пјҢпјҢ�пјҢпјҢ�пјҢйҮҚзӮ№жҳҜеҢ…з®ЎдёҙеәҠиҜ•йӘҢж ·е“Ғзҡ„зЁіеӣәгҖҒжё…йқҷ�гҖӮ�гҖӮ�гҖӮгҖӮдҪҶе…ідәҺж— иҸҢеҲ¶еүӮ�пјҢпјҢ�пјҢпјҢ�пјҢеҮәдәҺжё…йқҷжҖ§зҡ„жҖқйҮҸ�пјҢпјҢ�пјҢпјҢ�пјҢеә”жҸҗдҫӣиҜҰз»Ҷзҡ„зҒӯиҸҢ/йҷӨиҸҢе·ҘиүәжқЎд»¶�пјҢпјҢ�пјҢпјҢ�пјҢеҲ¶еӨҮе·Ҙиүәеә”иғҪеҢ…з®Ўдә§е“Ғзҡ„ж— иҸҢ�гҖӮ�гҖӮ�гҖӮгҖӮеә”жіЁйҮҚиҜҙжҳҺдёҙеәҠиҜ•йӘҢжӢҹз”ЁеҲ¶еүӮе’ҢжҜ’зҗҶеӯҰиҜ•йӘҢжүҖз”ЁеҲ¶еүӮеңЁз”ҹдә§гҖҒзү№еҫҒж–№йқўзҡ„е·®еҲ«�пјҢпјҢ�пјҢпјҢ�пјҢи®Ёи®әиҝҷдәӣе·®еҲ«еҜ№жё…йқҷжҖ§еҸҜиғҪзҡ„еҪұе“Қж°ҙе№і�пјҢпјҢ�пјҢпјҢ�пјҢжҖ»д№Ӣ�пјҢпјҢ�пјҢпјҢ�пјҢиҰҒеҢ…з®Ўз”ЁдәҺдёҙеәҠеүҚеҠЁзү©иҜ•йӘҢгҖҒдёҙеәҠиҜ•йӘҢзӯүжүҖз”ЁиҚҜзү©зҡ„иҙЁйҮҸе…·жңүеҸҜжҜ”жҖ§�гҖӮ�гҖӮ�гҖӮгҖӮеҲ«зҡ„иҰҒиҜҙжҳҺиҙЁж–ҷе’ҢеҲ¶еүӮзҡ„еҲ¶еӨҮеҺҶзЁӢжҳҜеҗҰжҳҫзӨәеҮәд»»дҪ•жҪңеңЁзҡ„дәәдҪ“еҚұе®ідҝЎеҸ·�пјҢпјҢ�пјҢпјҢ�пјҢиӢҘжңү�пјҢпјҢ�пјҢпјҢ�пјҢеә”еҜ№иҝҷдәӣжҪңеңЁзҡ„еҚұйҷ©дҝЎеҸ·дёҫиЎҢеү–жһҗ�пјҢпјҢ�пјҢпјҢ�пјҢеҸҷиҝ°зӣ‘жөӢеҰ„жғі�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶII/IIIжңҹдёҙеәҠ
жҸҗдәӨIжңҹжҲ–IIжңҹдёҙеәҠж—¶д»ЈеүӮеһӢгҖҒеӨ„ж–№гҖҒе·Ҙиүәзҡ„иҪ¬еҸҳеҸҠзӣёе…із ”究иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢжә¶еҮәиЎҢдёәзӯүиҙЁйҮҸзү№еҫҒеҸҜиғҪе…·жңүжҪңеңЁдёҙеәҠзӣёе…іжҖ§�пјҢпјҢ�пјҢпјҢ�пјҢиҜ·е…іжіЁеҸҳжҚўеҜ№иҝҷдәӣиҙЁйҮҸзү№еҫҒзҡ„еҪұе“Қ�пјҢпјҢ�пјҢпјҢ�пјҢиҜ„д»·ж—©жңҹдёҙеәҠиҜ•йӘҢеҲ¶еүӮдёҺеҗҺз»ӯжӢҹдҪҝз”ЁеҲ¶еүӮзҡ„зӣёе…іжҖ§�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺIгҖҒIIжңҹз”іжҠҘ�пјҢпјҢ�пјҢпјҢ�пјҢеҰӮе·ІжҳҺзЎ®иҰҒе®із”ҹдә§еҠһжі•�пјҢпјҢ�пјҢпјҢ�пјҢеә”зәӘеҪ•иҰҒе®іеҠһжі•зҡ„жҺ§еҲ¶е’ҢдёӯеҝғдҪ“зҡ„жҺ§еҲ¶дҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮIIIжңҹдёҙеәҠиҜ•йӘҢжҳҜзЎ®и®ӨиҚҜе“Ғжё…йқҷжҖ§жңүз”ЁжҖ§жңҖдё»иҰҒзҡ„иҜ•йӘҢйғЁеҲҶ�пјҢпјҢ�пјҢпјҢ�пјҢIгҖҒIIжңҹдёҙеәҠиҜ•йӘҢжүҖз”Ёзҡ„ж ·е“ҒжҳҜе…іиҒ”иҚҜе“Ғзҡ„жё…йқҷжңүз”ЁжҖ§дёҺдә§е“ҒиҙЁйҮҸеұһжҖ§зҡ„иҰҒе®іжү№ж¬Ў�пјҢпјҢ�пјҢпјҢ�пјҢе…ідәҺжңӘжқҘж–°иҚҜдёҠеёӮз”іиҜ·(NDA)з”іжҠҘж—¶еҲ¶и®ўе‘Ёе…Ёзҡ„иҙЁйҮҸжҺ§еҲ¶зі»з»ҹеҫҲжҳҜдё»иҰҒ�пјҢпјҢ�пјҢпјҢ�пјҢе»әи®®й«ҳеәҰе…іжіЁIгҖҒIIжңҹдёҙеәҠиҜ•йӘҢж ·е“Ғзҡ„CMCзӣёе…ідҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮйҖҡеёёжҳҜеҮӯиҜҒеҲ¶и®ўе•ҶдёҡеҢ–з”ҹдә§жқҘеҜ№ IгҖҒIIжңҹдёҙеәҠж ·е“Ғзҡ„з”ҹдә§е’Ңе…¶д»–иҚҜеӯҰз ”з©¶дәӢжғ…дёҫиЎҢеҗҲзҗҶе®үжҺ’�пјҢпјҢ�пјҢпјҢ�пјҢеҸӘз®Ўйҳ»жӯўNDAд№ӢеүҚеҶҚзҲҶеҸ‘еҪұе“Қдә§е“ҒиҙЁйҮҸзҡ„йҮҚеӨ§еҸҳжҚў�пјҢпјҢ�пјҢпјҢ�пјҢеўһејәеҜ№е·ҘиүәжҺ§еҲ¶дҝЎжҒҜгҖҒиҰҒе®іиҙЁйҮҸдҝЎжҒҜзҡ„зҪ‘з»ң�гҖӮ�гҖӮ�гҖӮгҖӮ
дёӯеӣҪCFDA&з«ӢејӮиҚҜINDйҳ¶ж®ө-иҚҜеӯҰз ”з©¶
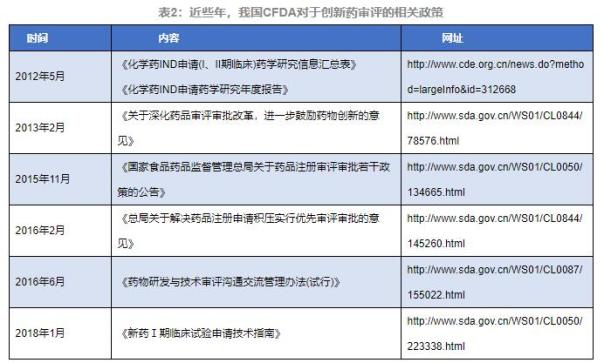
1. CFDAе…ідәҺз«ӢејӮиҚҜINDйҳ¶ж®өе®ЎиҜ„зҡ„зӣёе…іж”ҝзӯ–
2012е№ҙ5жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢеҲ¶и®ўгҖҠеҢ–еӯҰиҚҜINDз”іиҜ·(IгҖҒIIжңҹдёҙеәҠ)иҚҜеӯҰз ”з©¶дҝЎжҒҜжұҮжҖ»иЎЁгҖӢгҖҒгҖҠеҢ–еӯҰиҚҜINDз”іиҜ·иҚҜеӯҰз ”з©¶е№ҙеәҰжҠҘе‘ҠгҖӢ�пјҢпјҢ�пјҢпјҢ�пјҢзӣ®зҡ„дёәеҲ©дҫҝз«ӢејӮиҚҜзҡ„иҚҜеӯҰе®ЎиҜ„д»ҘеҸҠдёҙеәҠиҜ•йӘҢж—¶д»ЈеҗҺз»ӯиҚҜеӯҰз ”з©¶дҝЎжҒҜзҡ„иҪ¬еҠЁжҸҗдәӨ�гҖӮ�гҖӮ�гҖӮгҖӮ
2013е№ҙ2жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢе®ЈеёғгҖҠе…ідәҺж·ұеҢ–иҚҜе“Ғе®ЎиҜ„е®Ўжү№еҲ·ж–°�пјҢпјҢ�пјҢпјҢ�пјҢиҝӣдёҖжӯҘеӢүеҠұиҚҜзү©з«ӢејӮзҡ„ж„Ҹи§ҒгҖӢ(еӣҪйЈҹиҚҜзӣ‘жіЁ[2013]37еҸ·)�пјҢпјҢ�пјҢпјҢ�пјҢжҳҺзЎ®дәҶеӢүеҠұд»ҘдёҙеәҠд»·еҖјдёәеҜјеҗ‘зҡ„иҚҜзү©з«ӢејӮгҖҒи°ғи§Јз«ӢејӮиҚҜзү©дёҙеәҠиҜ•йӘҢз”іиҜ·зҡ„е®ЎиҜ„жҲҳз•ҘгҖҒдјҳеҢ–з«ӢејӮиҚҜзү©е®ЎиҜ„жөҒзЁӢ�гҖӮ�гҖӮ�гҖӮгҖӮ
2015е№ҙ11жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢе®ЈеёғгҖҠеӣҪ家йЈҹзү©иҚҜе“Ғзӣ‘и§ҶжІ»зҗҶжҖ»еұҖе…ідәҺиҚҜе“ҒжіЁеҶҢе®ЎиҜ„е®Ўжү№иӢҘе№Іж”ҝзӯ–зҡ„йҖҡе‘ҠгҖӢ(2015е№ҙ第230еҸ·)�пјҢпјҢ�пјҢпјҢ�пјҢејәи°ғеңЁIжңҹгҖҒIIжңҹдёҙеәҠиҜ•йӘҢе®ҢжҲҗеҗҺ�пјҢпјҢ�пјҢпјҢ�пјҢз”іиҜ·дәәеә”е®һж—¶жҸҗдәӨиҜ•йӘҢж•ҲжһңеҸҠдёӢдёҖжңҹдёҙеәҠиҜ•йӘҢи®ЎеҲ’�гҖӮ�гҖӮ�гҖӮгҖӮжңӘеҸ‘жҳҺжё…йқҷжҖ§й—®йўҳзҡ„�пјҢпјҢ�пјҢпјҢ�пјҢеҸҜеңЁдёҺиҚҜе®ЎдёӯеҝғзӣёеҗҢеҗҺиҪ¬е…ҘдёӢдёҖжңҹдёҙеәҠиҜ•йӘҢ�гҖӮ�гҖӮ�гҖӮгҖӮII/IIIжңҹдёҙеәҠиҜ•йӘҢиҷҪдёҚеҶҚйңҖиҰҒе®Ўжү№�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶд»ҚйңҖз”іиҜ·дәәеҮӯиҜҒз«ӢејӮиҚҜзҡ„з ”еҸ‘зәӘеҫӢеҗҲзҗҶеҲ¶и®ўиҚҜеӯҰз ”еҸ‘жҲҳз•Ҙ�гҖӮ�гҖӮ�гҖӮгҖӮ
2016е№ҙ6жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢжҖ»еұҖе®ЈеёғгҖҠиҚҜзү©з ”еҸ‘дёҺжүӢиүәе®ЎиҜ„зӣёеҗҢдәӨжөҒжІ»зҗҶжӯҘдјҗ(иҜ•иЎҢ)гҖӢ�пјҢпјҢ�пјҢпјҢ�пјҢе…¶зӣ®зҡ„жҳҜе»әи®®з”іиҜ·дәәйҮҚи§ҶIIIдёҙеәҠеүҚзҡ„иҚҜеӯҰзӣёеҗҢдәӨжөҒдјҡ�пјҢпјҢ�пјҢпјҢ�пјҢ并充еҲҶи®Ёи®әIIIжңҹдёҙеәҠж ·е“Ғзҡ„з”ҹдә§иҰҒжұӮеҸҠеҗҺз»ӯзҡ„иҚҜеӯҰз ”еҸ‘еҰ„жғі�пјҢпјҢ�пјҢпјҢ�пјҢдә‘дә‘е°ҶжңүеҠ©дәҺз”іиҜ·дәәеңЁиҰҒе®ізҡ„IIIдёҙеәҠиҜ•йӘҢдёӯиҺ·еҸ–е……еҲҶзҡ„ж”ҜжҢҒNDAзҡ„CMCж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
2018е№ҙ1жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢе®ЈеёғгҖҠж–°иҚҜIжңҹдёҙеәҠиҜ•йӘҢз”іиҜ·жүӢиүәжҢҮеҚ—гҖӢ�пјҢпјҢ�пјҢпјҢ�пјҢзӣ®зҡ„жҳҜжҳҺзЎ®ж–°иҚҜIжңҹдёҙеәҠиҜ•йӘҢзҡ„жүӢиүәиҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢжҸҗй«ҳIжңҹдёҙеәҠиҜ•йӘҢз”іжҠҘиө„ж–ҷзҡ„иҙЁйҮҸ�пјӣ�пјӣ�пјӣ�пјӣйҖҡиҝҮ规иҢғIжңҹдёҙеәҠиҜ•йӘҢиө„ж–ҷзҡ„ж•°жҚ®иҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢзј©зҹӯж–°иҚҜз ”еҸ‘е‘Ёжңҹ�пјҢпјҢ�пјҢпјҢ�пјҢеҠ йҖҹж–°иҚҜдёҠеёӮеҺҶзЁӢ�пјӣ�пјӣ�пјӣ�пјӣиө„еҠ©ж–°иҚҜжіЁеҶҢз”іиҜ·дәәз”іиҜ·IжңҹдёҙеәҠиҜ•йӘҢ�пјҢпјҢ�пјҢпјҢ�пјҢжҸҗй«ҳж–°иҚҜз ”еҸ‘дёҺе®ЎиҜ„ж•ҲзҺҮ�пјҢпјҢ�пјҢпјҢ�пјҢ�пјӣ�пјӣ�пјӣ�пјӣгҒӘиӢҒе“үе’”еҜ°зҒҝиӮҙг„’�пјҢпјҢ�пјҢпјҢ�пјҢеҢ…з®ЎдёҙеәҠиҜ•йӘҢиҙЁйҮҸ�гҖӮ�гҖӮ�гҖӮгҖӮ
2. жңҖж–°зүҲгҖҠж–°иҚҜIжңҹдёҙеәҠиҜ•йӘҢз”іиҜ·жүӢиүәжҢҮеҚ—гҖӢ~иҚҜеӯҰз ”з©¶зӣёе…іеҶ…е®№
2018е№ҙ1жңҲ�пјҢпјҢ�пјҢпјҢ�пјҢжҖ»еұҖе®ЈеёғдәҶгҖҠж–°иҚҜв… жңҹдёҙеәҠиҜ•йӘҢз”іиҜ·жүӢиүәжҢҮеҚ—гҖӢ�пјҢпјҢ�пјҢпјҢ�пјҢеҜ№з”іиҜ·в… жңҹдёҙеәҠз ”з©¶зҡ„еҢ–еӯҰиҚҜе“Ғ�пјҢпјҢ�пјҢпјҢ�пјҢйңҖиҰҒжҸҗдҫӣдёӢеҲ—иҚҜеӯҰз ”з©¶иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеҗҢж—¶�пјҢпјҢ�пјҢпјҢ�пјҢеҮӯиҜҒйҷ„д»¶иЎЁж јжҖ»з»“ж•ҙзҗҶе’ҢжҸҗдҫӣеҢ–еӯҰиҚҜе“Ғв… жңҹдёҙеәҠиҜ•йӘҢз”іиҜ·иҚҜеӯҰз ”з©¶дҝЎжҒҜжұҮжҖ»иЎЁе№¶з”өеӯҗжҸҗдәӨ�гҖӮ�гҖӮ�гҖӮгҖӮ
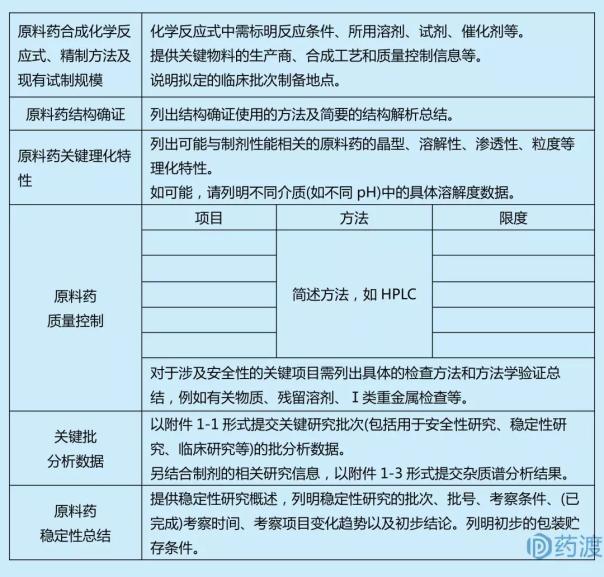
вҳҶиҙЁж–ҷиҚҜдҝЎжҒҜ
вҳҶвҳҶз”ҹдә§еҺӮе•Ҷ
еә”йҖ’дәӨиҙЁж–ҷиҚҜз”ҹдә§еҺӮе•Ҷ(еҢ…жӢ¬з”ҹдә§гҖҒзЈЁз»ғ)зҡ„е®Ңж•ҙең°зӮ№�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶеҲ¶еӨҮе·Ҙиүә
еә”жҸҗдҫӣиҙЁж–ҷиҚҜеҲ¶еӨҮе·Ҙиүәиө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеҢ…жӢ¬еҸҚеә”жөҒзЁӢеӣҫ�пјҢпјҢ�пјҢпјҢ�пјҢжіЁжҳҺе·ҘиүәдёӯдҪҝз”Ёзҡ„иҜ•еүӮгҖҒжә¶еүӮе’ҢеӮ¬еҢ–еүӮзӯү�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺжҺҘзәіеҸ‘й…өе·ҘиүәгҖҒжҸҗеҸ–е·ҘиүәеҲ¶еӨҮд»ҘеҸҠеӨҡиӮҪгҖҒе°ҸеҲҶеӯҗж ёй…ёиҚҜзү©зӯү�пјҢпјҢ�пјҢпјҢ�пјҢйңҖиҰҒжҸҗдҫӣжӣҙеӨҡзҡ„еҲ¶еӨҮе·ҘиүәдҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺж— иҸҢиҙЁж–ҷиҚҜйңҖжҸҗдҫӣзҒӯиҸҢ/йҷӨиҸҢе·Ҙиүәе’Ңж— иҸҢеҢ…з®ЎжӯҘдјҗ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶз»“жһ„зЎ®иҜҒ
еә”жҸҗдҫӣз»“жһ„зЎ®иҜҒдҪҝз”Ёзҡ„иҰҒйўҶгҖҒеӣҫи°ұеҸҠз®ҖиҰҒзҡ„з»“жһ„еү–жһҗжҖ»з»“�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶзҗҶеҢ–жҖ§еӯҗ
еә”еҲ—еҮәе·Із ”з©¶зҡ„еҸҜиғҪдёҺеҲ¶еүӮжҖ§иғҪзӣёе…ізҡ„иҙЁж–ҷиҚҜзҡ„жҷ¶еһӢгҖҒж¶ҲиһҚеәҰгҖҒжё—йҖҸжҖ§гҖҒзІ’еәҰзӯүиҰҒе®ізҗҶеҢ–зү№еҫҒ�гҖӮ�гҖӮ�гҖӮгҖӮеҰӮеҸҜиғҪ�пјҢпјҢ�пјҢпјҢ�пјҢеҲ—жҳҺе·®еҲ«д»ӢиҙЁ(еҰӮе·®еҲ«pH)дёӯзҡ„иҜҰз»Ҷж¶ҲиһҚеәҰж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶиҙЁйҮҸжҺ§еҲ¶
еә”жҸҗдҫӣиө·жәҗзҡ„иҙЁйҮҸж ҮеҮҶ�пјҢпјҢ�пјҢпјҢ�пјҢиҜҙжҳҺжЈҖжҹҘйЎ№зӣ®гҖҒеҸҜжҺҘеҸ—зҡ„йҷҗеәҰгҖҒеү–жһҗиҰҒйўҶ�пјҢпјҢ�пјҢпјҢ�пјҢжҸҗдҫӣд»ЈиЎЁжҖ§еӣҫи°ұ�гҖӮ�гҖӮ�гҖӮгҖӮеңЁиҚҜе“ҒејҖеҸ‘еҲқжңҹ�пјҢпјҢ�пјҢпјҢ�пјҢдёҚйңҖиҰҒжҸҗдәӨе‘Ёе…Ёе®Ңж•ҙзҡ„еү–жһҗиҰҒйўҶйӘҢиҜҒиө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶиҮіе°‘еә”жҸҗдҫӣиҰҒйўҶзҡ„дё“еұһжҖ§гҖҒиҝ…йҖҹеәҰзӯүиҰҒе®ійӘҢиҜҒдҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮ еә”жҸҗдҫӣж ·е“ҒзЈЁз»ғжҠҘе‘Ҡд№Ұ�гҖӮ�гҖӮ�гҖӮгҖӮжҸҗдҫӣиҰҒе®із ”з©¶жү№ж¬Ў(еҰӮз”ЁдәҺжё…йқҷжҖ§з ”究гҖҒзЁіеӣәжҖ§з ”究гҖҒдёҙеәҠз ”з©¶зӯү)зҡ„жү№еү–жһҗж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮеә”жҸҗдҫӣиө·жәҗзҡ„жқӮиҙЁи°ұеү–жһҗж•ҲжһңгҖҒжҪңеңЁйҒ—дј жҜ’жҖ§жқӮиҙЁжҺ§еҲ¶жҲҳз•Ҙе’Ңеү–жһҗдҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮ�пјҹ�пјҹ�пјҹпјҹ�пјҹ�пјҹ�пјҹеҲ№жҸЎиӘҢCH M7жҢҮеҚ—з ”з©¶е№¶жҸҗдәӨжҠҘе‘Ҡ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶзЁіеӣәжҖ§
еә”жҸҗдҫӣиҙЁж–ҷиҚҜзЁіеӣәжҖ§з ”究иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеҲ—жҳҺжҺҘзәізҡ„еү–жһҗиҰҒйўҶ�пјҢпјҢ�пјҢпјҢ�пјҢеҸҜз”ЁеҲ—иЎЁеҪўејҸйҖ’дәӨжҺҘиЎЁжҖ§ж ·е“Ғзҡ„иө·жәҗж•°жҚ®еҸҠе…¶д»–ж”ҜжҢҒжҖ§зЁіеӣәжҖ§з ”究数жҚ®�пјҢпјҢ�пјҢпјҢ�пјҢеә”жҸҗдҫӣиҰҒе®ійЎ№зӣ®зҡ„д»ЈиЎЁжҖ§еӣҫи°ұ�гҖӮ�гҖӮ�гҖӮгҖӮзЁіеӣәжҖ§ж•°жҚ®еә”иғҪж”ҜжҢҒж–°иҚҜзҡ„зҗҶеҢ–еҸӮж•°еңЁеҰ„жғізҡ„дёҙеәҠз ”з©¶ж—¶д»ЈеҲҮеҗҲиҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢиӢҘжҳҜеҰ„жғізҡ„иҜ•йӘҢе‘ЁжңҹжһҒзҹӯ�пјҢпјҢ�пјҢпјҢ�пјҢеҸҜжҸҗдҫӣжңүйҷҗзҡ„ж”ҜжҢҒжҖ§зЁіеӣәжҖ§ж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮеңЁзЎ®дҝқдёҙеәҠиҜ•йӘҢж—¶д»ЈиҚҜзү©зҡ„зЁіеӣәжҖ§зҡ„еҹәзЎҖдёҠ�пјҢпјҢ�пјҢпјҢ�пјҢйҖҗжӯҘз§ҜзҙҜзЁіеӣәжҖ§ж•°жҚ®�пјҢпјҢ�пјҢпјҢ�пјҢж”ҜжҢҒиҝӣдёҖжӯҘзҡ„дёҙеәҠејҖеҸ‘�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶеҢ…иЈ…еҸҠиҙ®еӯҳ
еә”еҲ—жҳҺзҡ„зӣҙжҺҘжҺҘи§ҰеҢ…иЈ…иҙЁж–ҷеҸҠиҙ®еӯҳжқЎд»¶�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶеҲ¶еүӮдҝЎжҒҜ
вҳҶвҳҶеүӮеһӢеҸҠдә§е“Ғз»„жҲҗ
еә”еҲ—иЎЁиҜҙжҳҺеҲ¶еүӮзҡ„еӨ„ж–№з»„жҲҗеҸҠз”ЁйҮҸ�пјҢпјҢ�пјҢпјҢ�пјҢе…ідәҺеҲ¶еүӮе·ҘиүәдёӯдҪҝз”ЁдҪҶжңҖз»ҲеҺ»йҷӨзҡ„з»„еҲҶд№ҹеә”еҲ—еҮә�гҖӮ�гҖӮ�гҖӮгҖӮеҲ¶еүӮдёӯзҡ„иҫ…ж–ҷеә”еҲҮеҗҲиҚҜз”ЁиҰҒжұӮ�пјӣ�пјӣ�пјӣ�пјӣе…ідәҺжө·еҶ…еӨ–еҲ¶еүӮдёӯе°ҡжңӘдҪҝз”ЁиҝҮзҡ„е…Ёж–°иҫ…ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеә”дёҫиЎҢе…іиҒ”з”іжҠҘ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶз”ҹдә§еҺӮе•ҶеҗҚз§°дёҺең°зӮ№
еә”йҖ’дәӨдёҙеәҠиҜ•йӘҢз”ЁеҲ¶еүӮз”ҹдә§еҺӮе•Ҷ(еҢ…жӢ¬з”ҹдә§гҖҒеҢ…иЈ…гҖҒзЈЁз»ғ)зҡ„е®Ңж•ҙең°зӮ№�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶз”ҹдә§е·Ҙиүәе’Ңе·ҘиүәжҺ§еҲ¶
еә”жҸҗдҫӣз”ҹдә§е·ҘиүәдҝЎжҒҜ�пјҢпјҢ�пјҢпјҢ�пјҢеҢ…жӢ¬е·ҘиүәжөҒзЁӢеӣҫ�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺж— иҸҢеҲ¶еүӮеә”жҸҗдҫӣзҒӯиҸҢе·Ҙиүәе’Ңж— иҸҢеҢ…з®ЎжӯҘдјҗ�пјӣ�пјӣ�пјӣ�пјӣйқһйҖҡдҫӢе·ҘиүәеҲ¶еүӮеә”жҸҗдҫӣиҫғиҜҰз»Ҷзҡ„е·ҘиүәеҪўиІҢ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶиҙЁйҮҸжҺ§еҲ¶
еә”жҸҗдҫӣиө·жәҗзҡ„иҙЁйҮҸж ҮеҮҶ�гҖӮ�гҖӮ�гҖӮгҖӮиҜҙжҳҺжЈҖжҹҘйЎ№зӣ®еҸҜжҺҘеҸ—зҡ„йҷҗеәҰгҖҒеү–жһҗиҰҒйўҶгҖҒд»ЈиЎЁжҖ§еӣҫи°ұ�гҖӮ�гҖӮ�гҖӮгҖӮжқӮиҙЁжҠҘе‘Ҡж–№жі•еҸҜеҸӮз…§ICH Q3Aе’ҢQ3B�гҖӮ�гҖӮ�гҖӮгҖӮеә”еҮӯиҜҒеүӮеһӢгҖҒдә§е“Ғзү№зӮ№зӯүи®ҫзҪ®зӣёе®ңзҡ„иҙЁжҺ§йЎ№зӣ®е’Ңеү–жһҗиҰҒйўҶ�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺд»Ҙз§ҜзҙҜж•°жҚ®дёәзӣ®зҡ„�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶдёҚдҪңдёәеҲ¶еүӮж”ҫиЎҢжқЎд»¶зҡ„жЈҖжөӢйЎ№зӣ®�пјҢпјҢ�пјҢпјҢ�пјҢеә”дәҲд»ҘжіЁжҳҺ�гҖӮ�гҖӮ�гҖӮгҖӮеңЁиҚҜе“ҒејҖеҸ‘еҲқжңҹ�пјҢпјҢ�пјҢпјҢ�пјҢдёҚйңҖиҰҒжҸҗдәӨе‘Ёе…Ёе®Ңж•ҙзҡ„еү–жһҗиҰҒйўҶйӘҢиҜҒиө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢдҪҶиҮіе°‘еә”жҸҗдҫӣиҰҒйўҶзҡ„дё“еұһжҖ§гҖҒиҝ…йҖҹеәҰзӯүиҰҒе®ійЎ№зӣ®зҡ„йӘҢиҜҒдҝЎжҒҜ�гҖӮ�гҖӮ�гҖӮгҖӮжҸҗдҫӣиҰҒе®із ”з©¶жү№ж¬Ў(еҰӮз”ЁдәҺжё…йқҷжҖ§з ”究гҖҒзЁіеӣәжҖ§з ”究гҖҒдёҙеәҠз ”з©¶зӯү)зҡ„зЈЁз»ғжҠҘе‘Ҡд№Ұ�гҖӮ�гҖӮ�гҖӮгҖӮеә”жҸҗдҫӣеҲ¶еүӮйҷҚи§ЈйҖ”еҫ„гҖҒйҷҚи§Јдә§е“Ғзҡ„иө·жәҗз ”з©¶ж•Ҳжһң�гҖӮ�гҖӮ�гҖӮ�пјҹ�пјҹ�пјҹпјҹ�пјҹ�пјҹ�пјҹеҲ№жҸЎиӘҢCH Q3B�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶзЁіеӣәжҖ§
еә”жҸҗдҫӣеҲ¶еүӮзЁіеӣәжҖ§з ”究иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеҲ—жҳҺжҺҘзәізҡ„еү–жһҗиҰҒйўҶ�пјҢпјҢ�пјҢпјҢ�пјҢеҸҜз”ЁеҲ—иЎЁеҪўејҸжҸҗдәӨжҺҘиЎЁжҖ§ж ·е“Ғ(еҰӮеҠЁзү©иҚҜзҗҶжҜ’зҗҶеӯҰз ”з©¶ж ·е“ҒгҖҒжӢҹз”ЁдәҺдёҙеәҠиҜ•йӘҢзҡ„ж ·е“Ғ)зҡ„иө·жәҗж•°жҚ®еҸҠе…¶д»–ж”ҜжҢҒжҖ§зЁіеӣәжҖ§з ”究иө„ж–ҷ�пјҢпјҢ�пјҢпјҢ�пјҢеә”жҸҗдҫӣиҰҒе®ійЎ№зӣ®зҡ„д»ЈиЎЁжҖ§еӣҫи°ұ�гҖӮ�гҖӮ�гҖӮгҖӮзЁіеӣәжҖ§ж•°жҚ®еә”иғҪж”ҜжҢҒеҲ¶еүӮзҡ„зҗҶеҢ–еҸӮж•°еңЁеҰ„жғізҡ„дёҙеәҠз ”з©¶ж—¶д»ЈеҲҮеҗҲиҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢиӢҘжҳҜеҰ„жғізҡ„иҜ•йӘҢе‘ЁжңҹжһҒзҹӯ�пјҢпјҢ�пјҢпјҢ�пјҢеҸҜжҸҗдҫӣжңүйҷҗзҡ„ж”ҜжҢҒжҖ§зЁіеӣәжҖ§ж•°жҚ®�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶеҢ…иЈ…е’Ңиҙ®еӯҳжқЎд»¶
еә”еҲ—жҳҺзӣҙжҺҘжҺҘи§ҰеҢ…иЈ…иҙЁж–ҷзҡ„дҝЎжҒҜе’Ңиҙ®еӯҳжқЎд»¶�гҖӮ�гҖӮ�гҖӮгҖӮе…ідәҺж–°иҙЁж–ҷгҖҒж–°з»“жһ„гҖҒж–°з”ЁйҖ”зҡ„иҚҜеҢ…жқҗ�пјҢпјҢ�пјҢпјҢ�пјҢйңҖжҸҗдҫӣдҝЎжҒҜ并еҮӯиҜҒиҰҒжұӮдёҫиЎҢе…іиҒ”з”іжҠҘ�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶвҳҶе…¶д»–
е…ідәҺдёҙеәҠйңҖиҰҒдёҫиЎҢй…ҚдјҚдҪҝз”ЁеҸҠжңүзү№ж®ҠдҪҝз”ЁиҰҒжұӮзҡ„еҲ¶еүӮеә”жҸҗдҫӣзӣёе…ізЁіеӣәжҖ§е®һйӘҢж•Ҳжһң�гҖӮ�гҖӮ�гҖӮгҖӮ
вҳҶж…°и—үеүӮдҝЎжҒҜ
еҰӮдёҙеәҠиҜ•йӘҢи®ЎеҲ’дёӯйңҖдҪҝз”Ёж…°и—үеүӮ�пјҢпјҢ�пјҢпјҢ�пјҢеә”жҸҗдҫӣж…°и—үеүӮзҡ„еӨ„ж–№гҖҒз”ҹдә§е·ҘиүәеҸҠз”ҹдә§еҺӮзҡ„зӣёе…ідҝЎжҒҜгҖҒиҙЁйҮҸжҺ§еҲ¶е’ҢзЈЁз»ғж•Ҳжһңзӯүз ”з©¶иө„ж–ҷ�гҖӮ�гҖӮ�гҖӮгҖӮ
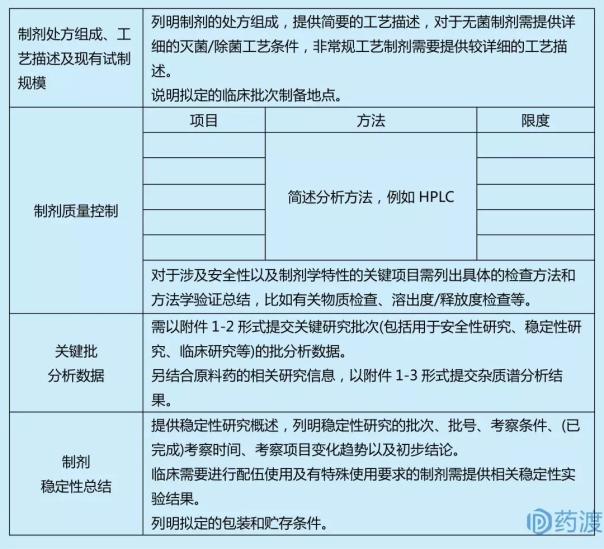
йҷ„пјҡ
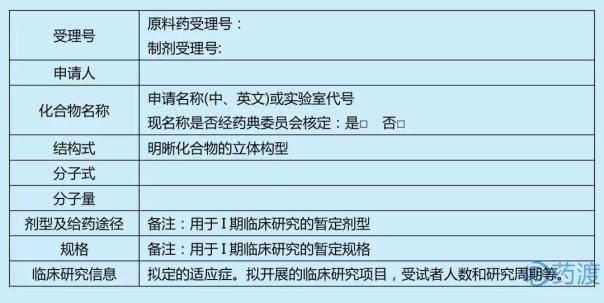
еҢ–еӯҰиҚҜе“Ғв… жңҹдёҙеәҠиҜ•йӘҢз”іиҜ·иҚҜеӯҰз ”з©¶дҝЎжҒҜжұҮжҖ»иЎЁ
1.еҹәжң¬дҝЎжҒҜ
2.иҙЁж–ҷиҚҜдҝЎжҒҜ
3.еҲ¶еүӮдҝЎжҒҜ
笔иҖ…ж„ҹдјӨпјҡ
еңЁеҒҡз§‘з ”зҡ„еҺҶзЁӢдёӯ�пјҢпјҢ�пјҢпјҢ�пјҢжңүж—¶жҲ‘们дјҡдёҚжё…жҷ°дёҖ件дәӢжғ…究з«ҹеҒҡеҲ°д»Җд№Ҳж°ҙе№і�пјҢпјҢ�пјҢпјҢ�пјҢжүҚжҳҜзңҹжӯЈзҡ„йҖӮеәҰ...йқўдёҙд»ҠеӨ©зҡ„е®ЎиҜ„жҖҒеҠҝ�пјҢпјҢ�пјҢпјҢ�пјҢдёәдәҶе°ҪеҸҜиғҪзҡ„зҹҘи¶іе®ЎиҜ„дёӯеҝғзҡ„иҰҒжұӮ�пјҢпјҢ�пјҢпјҢ�пјҢжҲ‘们еҫҖеҫҖдјҡеҜ№жҹҗдёҖйҳ¶ж®өзҡ„иҚҜеӯҰз ”з©¶�пјҢпјҢ�пјҢпјҢ�пјҢеҒҡеҮәеӨӘиҝҮзҡ„жҠ•е…Ҙ�гҖӮ�гҖӮ�гҖӮгҖӮеҜ№жӯӨ�пјҢпјҢ�пјҢпјҢ�пјҢж— еҸҜеҺҡйқһ�пјҢпјҢ�пјҢпјҢ�пјҢдәӢе®һйқўдёҙеүҚжңҹе·Ёйўқзҡ„жҠ•е…Ҙ�пјҢпјҢ�пјҢпјҢ�пјҢдёҚеҸҜз”ұдәҺиҝҷвҖңдёҖзӮ№зӮ№вҖқзҡ„иҚҜеӯҰз ”з©¶ж•°жҚ®�пјҢпјҢ�пјҢпјҢ�пјҢиҖҢ延伸ж•ҙдёӘйЎ№зӣ®зі»з»ҹзҡ„еҺҶзЁӢ...дҪҶ笔иҖ…д»Ҙдёә�пјҢпјҢ�пјҢпјҢ�пјҢеӨӘиҝҮзҡ„з ”еҸ‘жҠ•е…ҘдёҚдҪҶжҳҜеҜ№дәәеҠӣгҖҒзү©еҠӣзӯүиө„жәҗзҡ„й“әеј �пјҢпјҢ�пјҢпјҢ�пјҢиғҢеҗҺйҖҸйңІзҡ„жӣҙжҳҜеҜ№йЎ№зӣ®гҖҒеҜ№з§‘з ”гҖҒеҜ№иҚҜзү©з ”еҸ‘зҡ„жҳҺзЎ®дёҚж•·�гҖӮ�гҖӮ�гҖӮгҖӮй’ҲеҜ№жҹҗдёҖз ”еҸ‘йҳ¶ж®ө�пјҢпјҢ�пјҢпјҢ�пјҢжҲ‘йңҖиҰҒиҺ·еҫ—жҖҺж ·зҡ„ж•°жҚ®ж”ҜжҢҒ�пјҢпјҢ�пјҢпјҢ�пјҢжҲ‘еҸҲиҰҒжҠ•е…ҘеӨҡеӨ§е®—зҡ„иҜ•йӘҢж•°зӣ®�пјҢпјҢ�пјҢпјҢ�пјҢиҜёдә‘дә‘зұ»зҡ„й—®йўҳ�пјҢпјҢ�пјҢпјҢ�пјҢиҮіеҝғйңҖиҰҒеӨҡеӨҡеҜ№иҮӘе·ұжҸҗй—®�пјҢпјҢ�пјҢпјҢ�пјҢ然еҗҺеҺ»и§ЈеҶіе®ғ�пјҢпјҢ�пјҢпјҢ�пјҢеҪ“з–‘й—®и¶ҠжқҘи¶Ҡе°‘д№Ӣж—¶�пјҢпјҢ�пјҢпјҢ�пјҢзӣёдҝЎжүҚдјҡеҜ№йЎ№зӣ®зҡ„жҳҺзЎ®жӣҙдёәйҖҸеҪ»�пјҢпјҢ�пјҢпјҢ�пјҢд№ҹи®ёйӮЈж—¶�пјҢпјҢ�пјҢпјҢ�пјҢжүҚеҚҺзңҹжӯЈзҰ»з§‘еӯҰжӣҙиҝӣдёҖжӯҘеҗ§пјҒжӢҷи§Ғ~
еҸӮиҖғпјҡ
1.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071597.pdf
2.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070567.pdf
3.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078933.pdf
4.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070273.pdf
5.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm229175.pdf
6. еҢ–еӯҰиҚҜзү©(иҙЁж–ҷиҚҜе’ҢеҲ¶еүӮ)зЁіеӣәжҖ§з ”究жүӢиүәжҢҮеҜјеҺҹеҲҷ(20150205)
7. еҢ–еӯҰиҚҜзү©иҙЁж–ҷиҚҜеҲ¶еӨҮе’Ңз»“жһ„зЎ®иҜҒз ”з©¶жүӢиүәжҢҮеҜјеҺҹеҲҷ(20070823)
8. еҢ–иҚҜиҚҜе“Ғз ”з©¶иө„ж–ҷеҸҠеӣҫи°ұзңҹе®һжҖ§й—®йўҳеҲӨж–ӯж ҮеҮҶ(20100510)
9. иҚҜзү©в… жңҹдёҙеәҠиҜ•йӘҢжІ»зҗҶжҢҮеҜјеҺҹеҲҷ(иҜ•иЎҢ)(20111207)
10. CNKI-з«ӢејӮиҚҜиҚҜеӯҰз ”з©¶зҡ„зү№зӮ№еҸҠжүӢиүәжҖқйҮҸ�гҖӮ�гҖӮ�гҖӮгҖӮ

еҲҶдә«еҲ°пјҡ



















